
殷世荣主任医生介绍,都是侧索硬化症,原发性和肌萎缩是一种吗?
原发性侧索硬化症和肌萎缩侧索硬化症是不是同一种病?先说结果,这两种疾病并不是一种病,虽然名称相似,症状也有部分重叠,但本质上是两种不同的运动神经元病。殷世荣主任从病因、症状、进展三维度,为大家揭开它们的区别。

一、病因与发病机制
原发性侧索硬化症的病因至今未明,但研究提示可能与遗传、氧化应激、免疫异常及环境毒素有关。约5%-10%的患者有家族史,常染色体显性遗传模式较为常见,基因突变可能通过影响神经元代谢或抗氧化能力引发疾病。
肌萎缩侧索硬化症的病因更为复杂,是遗传与环境共同作用的结果。20%的家族性患者存在基因突变,如超氧化物歧化酶1基因突变会导致神经元内毒性蛋白堆积,引发细胞死亡。散发性ALS则与谷氨酸兴奋性毒性、线粒体功能障碍、神经炎症等机制相关。
二、症状表现
原发性侧索硬化症的核心特征是上运动神经元损伤,表现为:
痉挛性瘫痪:双下肢对称性僵硬、乏力,行走呈剪刀步态,随病情进展累及上肢,出现手指运动迟缓、书写困难。
假性延髓麻痹:晚期出现吞咽困难、饮水呛咳、构音障碍及强哭强笑等情绪失控表现。
无肌肉萎缩:与肌萎缩侧索硬化症不同,原发性侧索硬化症患者肌肉体积通常正常,仅在疾病晚期因长期废用可能出现轻度萎缩。
肌萎缩侧索硬化症则同时累及上、下运动神经元,症状更为多样:
肌肉无力与萎缩:症状常为手指活动笨拙、无力,逐渐蔓延至手臂、肩部及下肢,肌肉萎缩呈鹰爪手或倒三角腿形态。
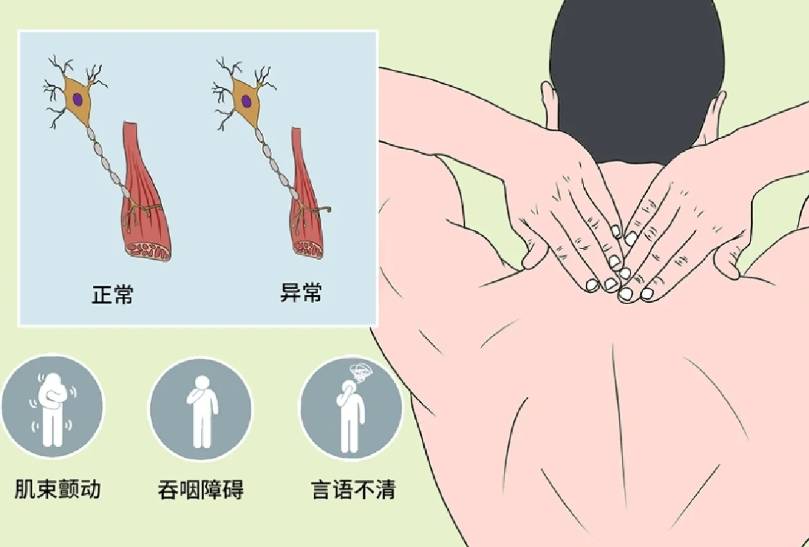
肌束颤动:受累肌肉可见肉眼可见的跳动,是下运动神经元损伤的标志性表现。
延髓症状:约30%的患者以吞咽困难、构音障碍起病,后期可出现呼吸肌无力,需依赖呼吸机维持生命。
三、疾病进展
原发性侧索硬化症的进展速度显著慢于肌萎缩侧索硬化症。患者从下肢症状出现到累及上肢通常需5-10年,部分患者发病20年后仍能独立行走。而肌萎缩侧索硬化症的病程呈暴发性特征,平均生存期仅3-5年,从手指无力到全身瘫痪、呼吸衰竭的进展可能仅需1-2年。
这种差异与神经元损伤范围密切相关。原发性侧索硬化症仅影响皮质脊髓束,而肌萎缩侧索硬化症会同时破坏皮质脊髓束、脊髓前角细胞及脑干运动神经核,导致更广泛的神经功能丧失。