
中国超 80% 癌症新药临床试验有缺陷?北大团队最新顶刊
中国的癌症新药临床试验,超八成都有缺陷?
这是本月柳叶刀子刊 The Lancet Oncology 发表的一篇重磅研究,由北京大学、耶鲁大学、哈佛大学等研究团队共同完成。研究系统评估了中国国家药品监督管理局(NMPA)药品审评中心(CDE)批准的癌症药物临床试验的证据质量,得出了这样的结论——
2017~2021 年,中国批准上市的癌症新药所涉及的 205 项关键性研究中,高达 82% 存在设计、实施或报告方面的缺陷。[1]
研究同时还指出,这不仅是中国,也是全球新药研究的通病。
图源:参考资料 1
中国癌症新药临床试验,82% 存在缺陷
先来详细了解一下研究到底得出了哪些结论。
研究团队根据 NMPA 的公开数据,共分析了 2017~2021 年期间,中国批准上市的 77 种癌症新药的 86 个适应症、62 个补充适应症所涉及的 205 项研究。
主要评价内容为「支持关键性研究的强度」,通过研究设计和研究质量两个方面来衡量。
研究设计:随机、单臂;
研究质量:随机对照试验(RCT)的偏倚风险、单臂试验是否采用外部对照臂并调整混杂因素。
图源:参考资料 1
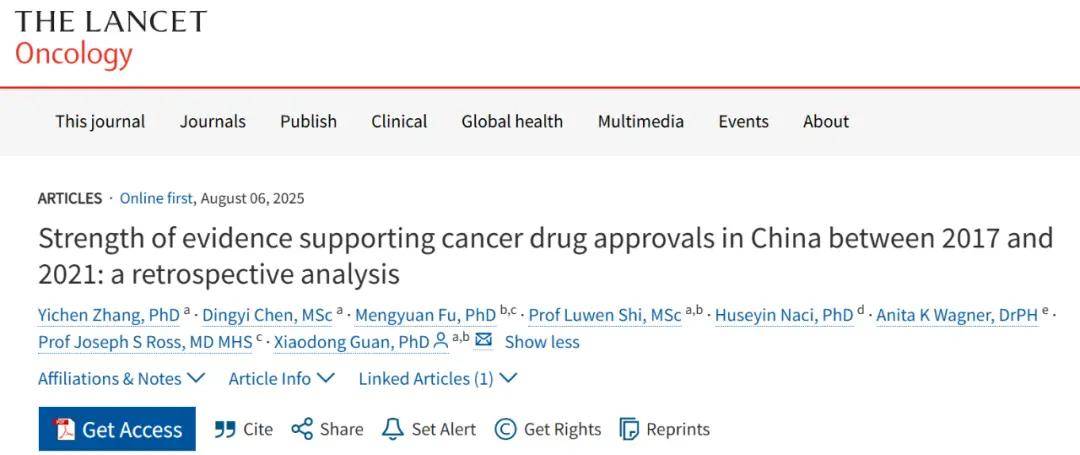
结果显示,在 148 个适应症批准中,30%(44 项)仅以单臂试验作为依据,其中 11% 使用早期试验的汇总数据作为外部对照,且未调整混杂因素。
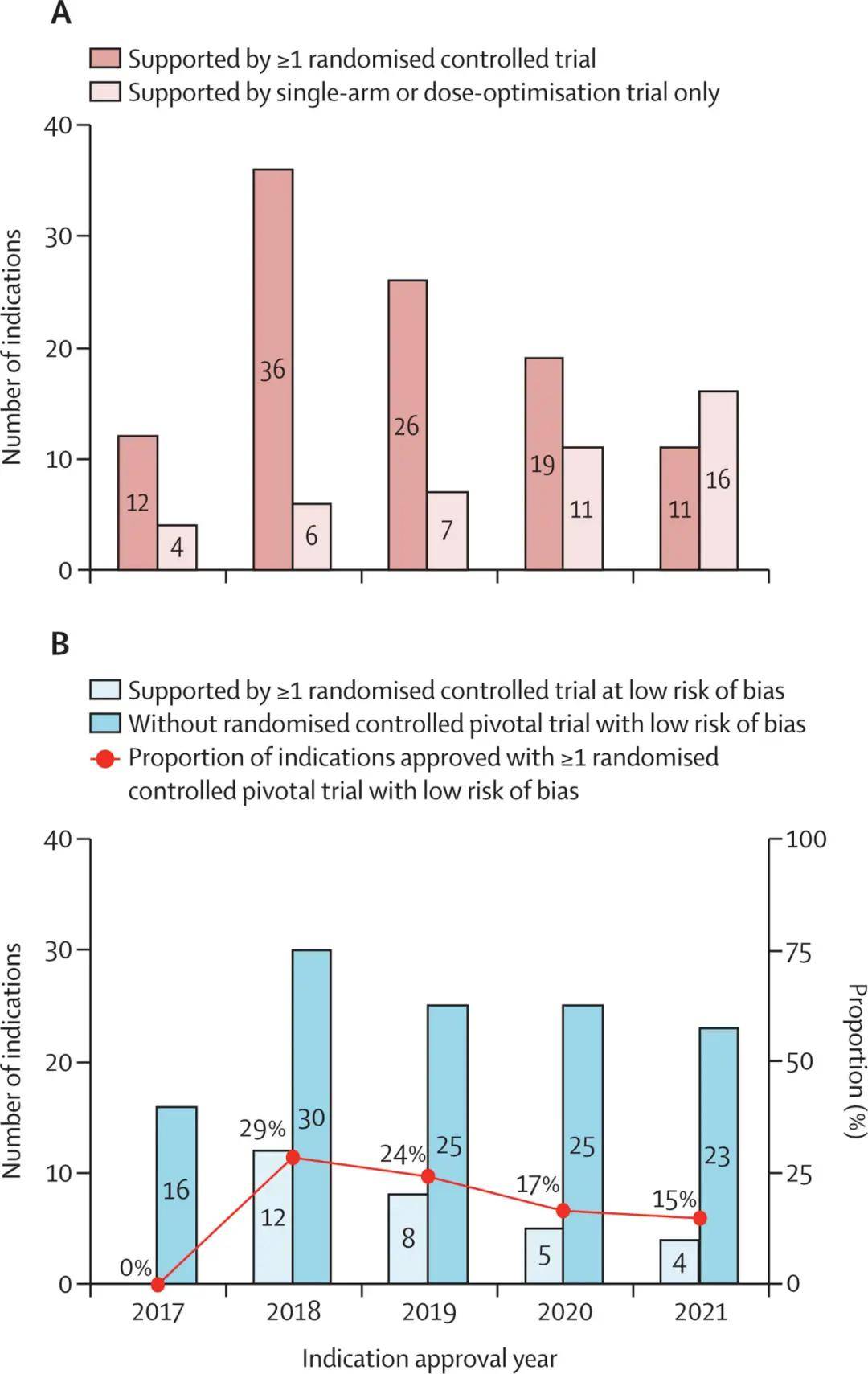
在 128 项可评估的 RCT 中,38%(48 项)存在高风险偏倚,主要源于结局数据缺失(46 项)和随机化缺陷(5 项)。37%(47 项)存在「部分担忧」,主要因未设盲(33 项)和选择性报告(22 项)。仅 26%(33 项)为低风险偏倚。
而且,我们普遍认为更有说服力、更「高级」的国际多中心试验,偏倚风险显著高于区域试验(80% vs 63%)。
总体而言,在全部 182 项可评估质量的关键试验中,有高达 82%(149 项)在偏倚控制方面存在局限性。研究团队指出:「高偏倚风险会直接导致疗效估计失真,缺失数据偏倚会低估效应量,随机化缺陷则会显著高估效应量。」[1]
图源:参考资料 1
这项研究的另一个结论则是,新药临床试验的终点不够「硬」。
通常认为,除非药物的临床需求非常紧迫,大多数新药应该使用明确改善临床预后的「硬终点」。但研究所得的分析结果,却和我们想象的不同:
在这些新药试验中,高达 63% 的 RCT(77/122)并不以总生存期(OS)为主要终点,而是使用包括无进展生存期(PFS)在内的指标作为替代终点。
研究团队指出,当替代终点与 OS 的相关性未经验证时,可能夸大临床获益,尤其在偏倚风险高的试验中更明显。
研究团队分析认为,替代终点的「滥用」,一方面是试验设计的问题,另一方面也是相关监管部门在审批时的权衡。
癌症患者迫切的治疗需求,促使癌症药物在审批时常常是走「加速审批」通道,而想要实现加速审批,在证据质量上免不了要进行妥协,其中的度,非常不好把握。
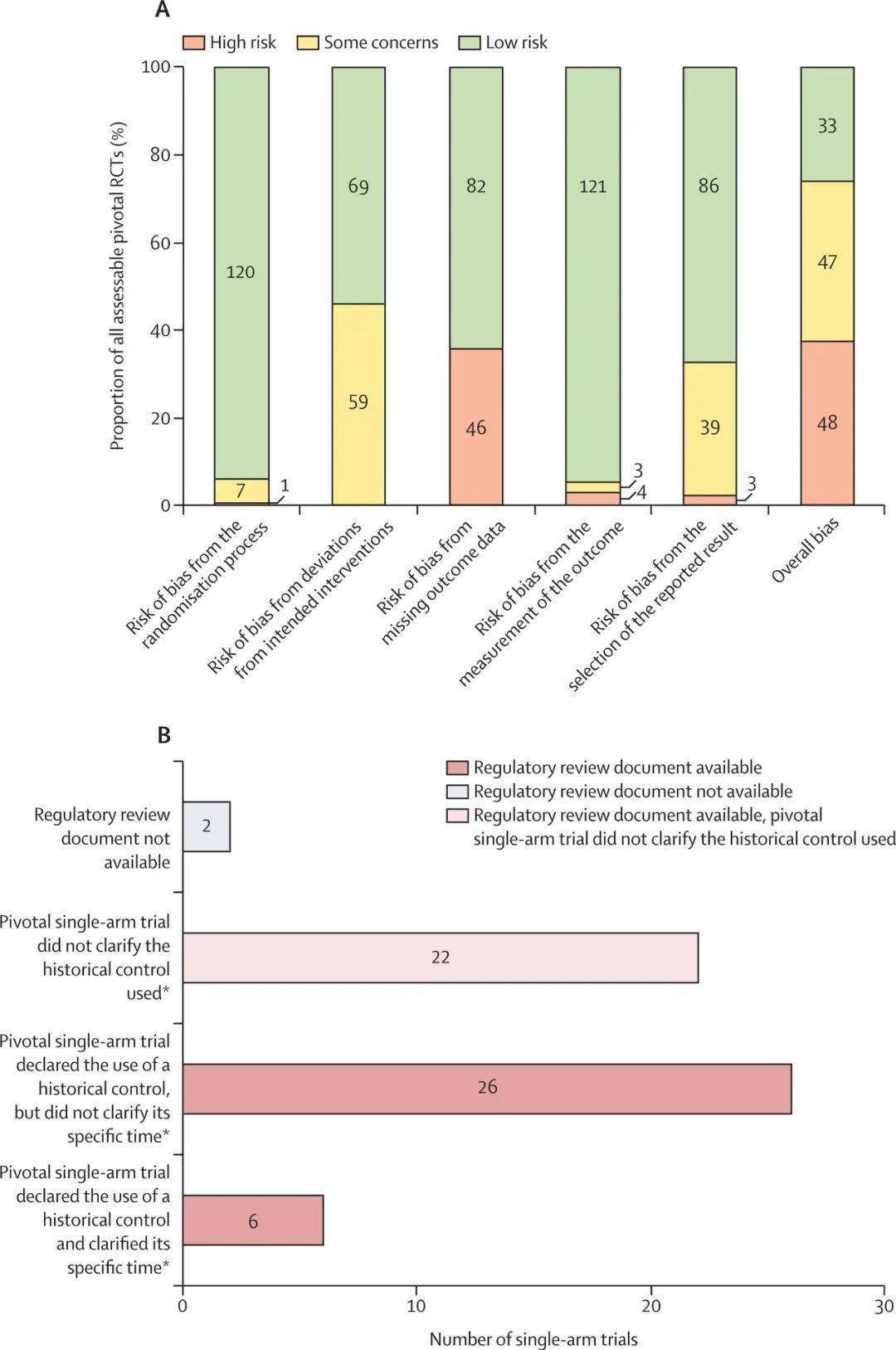
而除了审批的宽松度,透明度或许也存在问题。上述研究显示,33% 的 RCT 协议未公开,单臂试验混杂调整方法均未披露。
「公开评审细节和原始数据,是减少选择性报告的关键。」研究团队总结道。[1]
「问题试验」或将产生 9 倍临床影响,专家直言:警惕「伪创新」
「82%」这一数字虽然惊人,却与全球临床试验质量的总体趋势并无二致。
2019 年,英国伦敦政治经济学院研究团队曾在 BMJ 发表研究论文,对 2014~2016 年欧洲药物管理局(EMA)批准的癌症药物的随机对照试验进行分析。
结果显示,EMA 在此期间批准的 32 种癌症新药所涉及的 54 项关键研究中,76%(41 项)为 RCT,24%(13 项)为非随机研究或单臂研究。其中,49%(19 项)RCT 被判定为「存在偏倚高风险」。
而且,只有 26%(10 项)RCT 将 OS 作为主要终点,其余都用了替代指标。[2]

图源:参考资料 2
看完欧洲,再来看看美国。
开头所提研究的团队之一,耶鲁大学医学院的研究团队曾在 JAMA Network Open 上发表论文,详细分析了 1995~1997 年、2005~2007 年以及 2015~2017 年间获得美国 FDA 批准的 273 个新药和生物制品。
结果发现,只基于 1 项关键性试验就获批的适应症占比明显增加,从 1995~1997 年的 4.7%,上升至 2005~2007 年的 12.7%,到 2015~2017 年已经达到 17.0%。至少有 2 项关键性试验支持的占比例则相应地从 80.6% 降至 52.8%。[3]
此外,另一个瑞士团队分析了 2009~2021 年间,所有获得美国 FDA 批准的「头对头」RCT,结果显示,高达 55% 的试验采用了有利于试验组的方案规则。[4]
基于这一结果,瑞士团队直接质问:「新药是否真的优于旧药?」[4]


图源:参考资料 3、4
类似的质问并非孤例,加拿大皇后大学肿瘤学专家 Bishal Gyawali 教授也曾公开直言,需要警惕新药研发和试验中的「伪创新」。[5]
他以胰腺癌为例,指出:「25 年创新研发,患者的临床收益却微乎其微。部分新药实际上只是『昂贵版旧药』。」[5]
今年 5 月,海军军医大学第三附属医院研究团队在 BMJ 发表论文,构建了一个完整的「循证医学证据污染链」。
研究团队将设计、实施和分析等步骤存在问题的试验称为「问题试验」,通过深入追踪 1300 项问题试验,研究团队发现:平均每项问题试验,会污染 3 项循证医学研究,而每项循证医学研究又会进一步污染 3 项国际临床诊疗指南。
换句话说,临床试验不够严格,对患者的影响将成倍放大,从「问题试验」到「问题结论」,通过循证医学证据链,临床影响或将放大 9 倍。[6]

图源:参考资料 6
对此,研究者及同期述评文章建议:「临床医生应该有意识地溯源评估证据来源的可靠性,避免过度依赖 P 值进行临床决策。」[6]
但这对于临床实践而言,无疑是额外的负担。
新药获批究竟需要怎样质量的研究证据支持,才能在满足临床需求,和保证疗效与安全性之间找到最佳平衡,是全球监管部门共同的课题。
策划:肯德羊|监制:islay
题图来源:CCTV